Energy Frameworks
Energy frameworks arose out of a desire to better understand crystal packing by combining efficient calculation of intermolecular interaction energies with a novel graphical representation of their magnitude.
In this manner intriguing questions, such as why some crystals bend with an applied force while others break, and why one polymorph of a drug exhibits exceptional tabletability compared to others, can be addressed in terms of the anisotropy of the topology of pairwise intermolecular interaction energies. This approach was described and applied to a sample of organic molecular crystals with known bending, shearing and brittle behaviour, to illustrate its use in rationalising their mechanical behaviour at a molecular level.
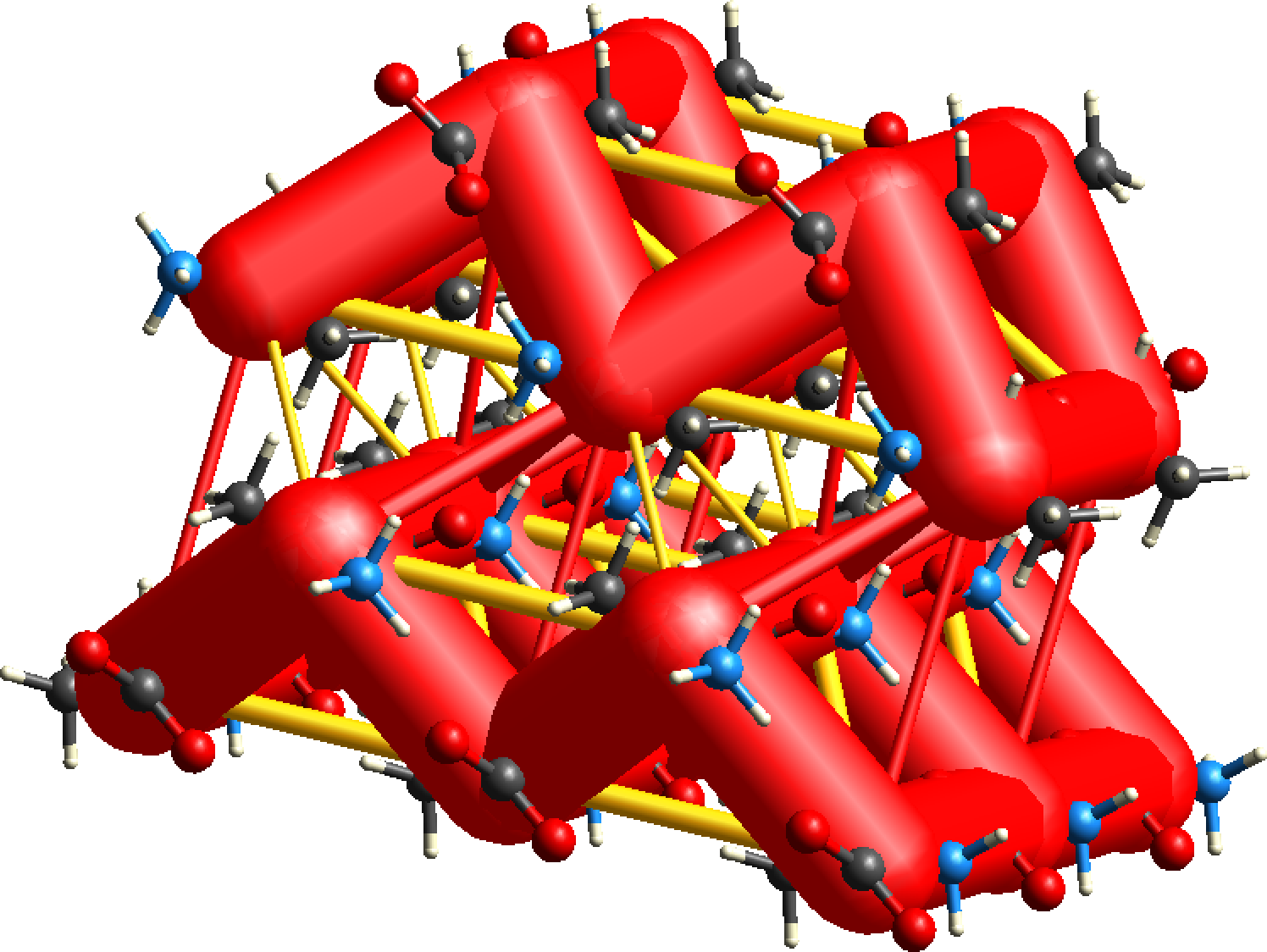
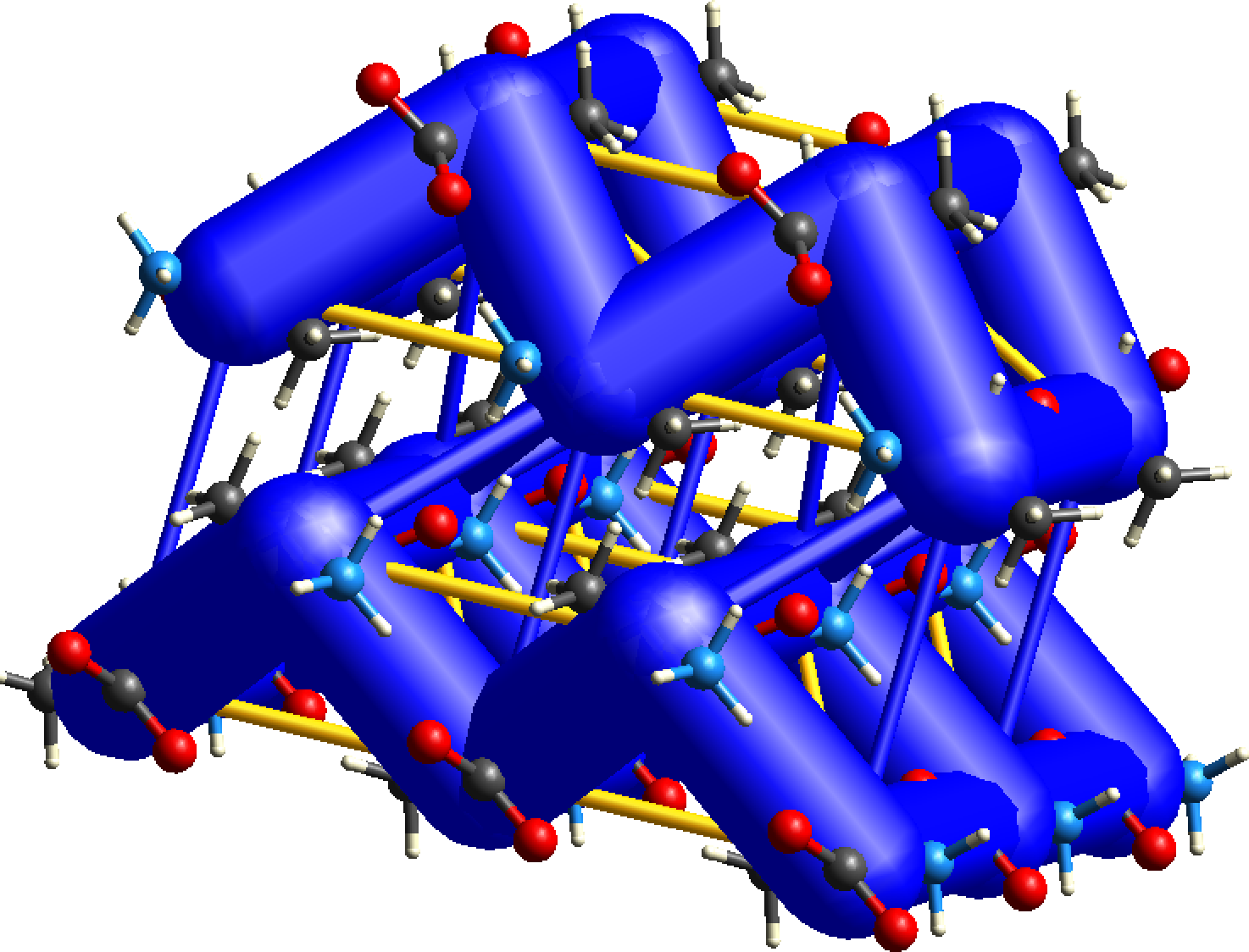
CrystalExplorer interaction energies underpin the calculation of energy frameworks. Energies between molecular pairs are represented as cylinders joining the centroids of pairs of molecules, with the cylinder radius proportional to the magnitude of the interaction energy. Frameworks can be constructed for (red cylinders), (green) and (blue).
Energy frameworks offer a powerful and unique way to visualize the supramolecular architecture of molecular crystal structures. The cylinders that make up the framework represent the relative strengths of molecular packing in different directions. An overall scale factor can be used to expand or contract the framework cylinders, meaning energy frameworks are only directly comparable if this scale factor is the same. To avoid crowded diagrams, weaker interactions below a certain energy threshold can be omitted, and because of this the absence of cylinders along a particular direction in the energy framework does not necessarily imply the complete absence of any stabilizing interaction in that direction. Judicious selection of this threshold is needed.
More recently we have allowed for the framework diagrams to include destabilizing (i.e. positive) interaction energies as well, and these are depicted as yellow cylinders, and only for the electrostatic and total energies.
Constructing Energy Frameworks
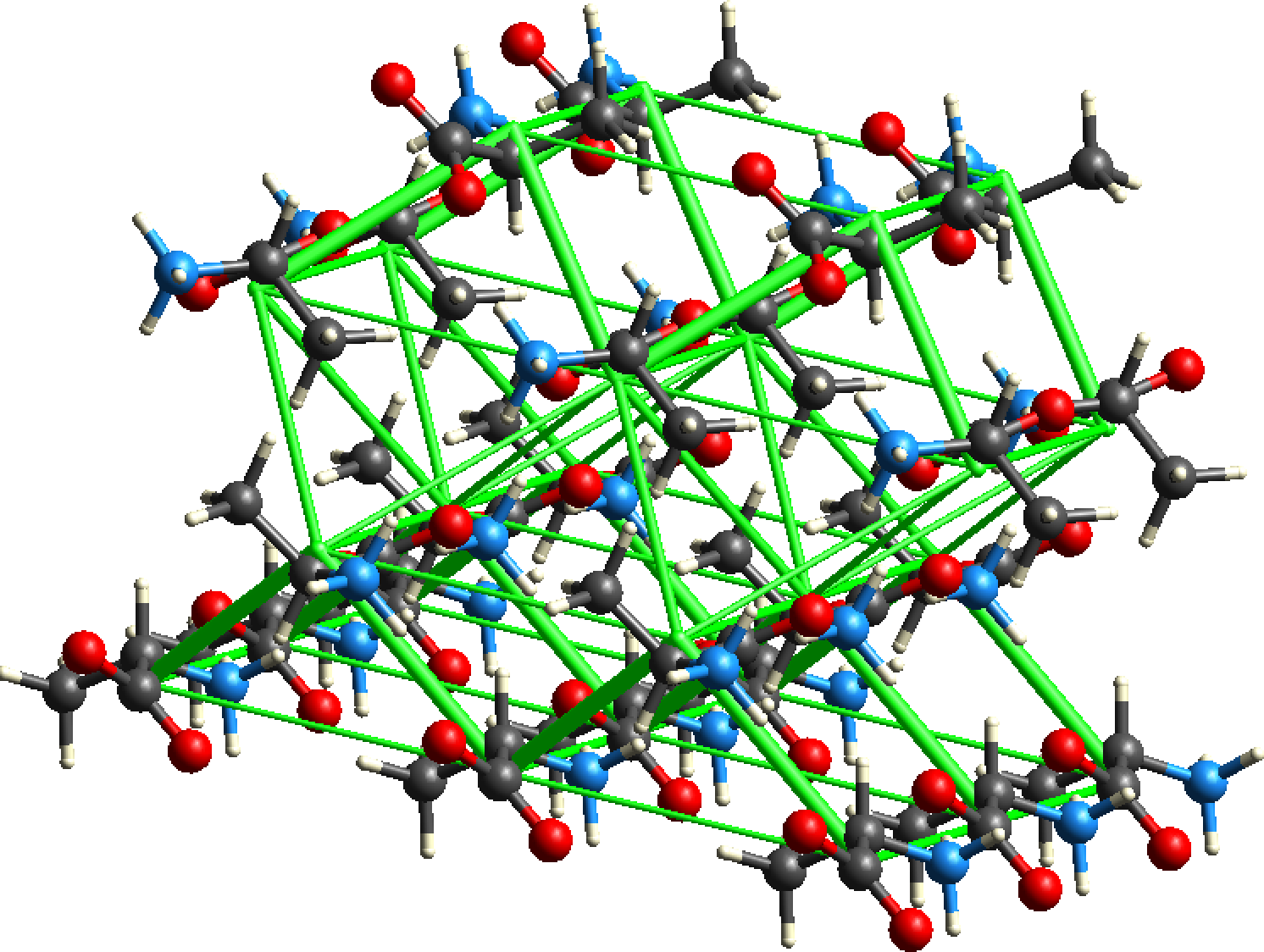
- It is a (pretty obvious) prerequisite that interaction energies for a cluster of molecules (usually the first coordination sphere - nearest neighbours) must be calculated first.
- Close the Information dialog (containing the list of energies, etc.). Then construct a cluster of interest,
typically with the Generate Unit Cells
 button, making
ure that all molecule fragments are completed (e.g. using the Complete Fragments
button, making
ure that all molecule fragments are completed (e.g. using the Complete Fragments  button).
button). - Selecting Display > Energy Frameworks... (
Ctrl-For⌘-Fon Mac) will open the Energy Frameworks dialog. This allows you to toggle between Coulomb Energy, Dispersion Energy and Total Energy frameworks in the graphics window.
The energy frameworks below for L-alanine show a cluster of unit cells. The CE-B3LYP model has been used, and the scale for tube/cylinder size is 80, and energies of magnitude smaller than 5 kJ/mol have been omitted for clarity. Notice the dominance of electrostatic energies in this case (relative to the dispersion term), and the importance of depicting the destabilizing energies (yellow cylinders).